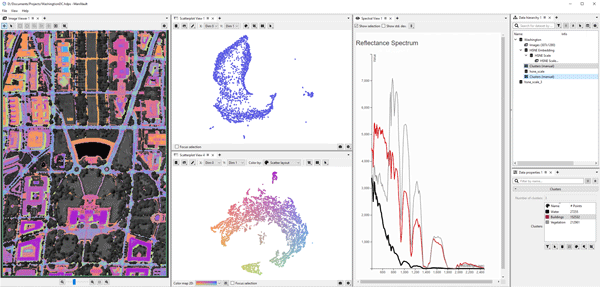
Exploration and analysis of high-dimensional data are important tasks in many fields that produce large and complex data, like the financial sector, systems biology, or cultural heritage. Tailor-made visual analytics...

I am a professor for visualization in the Vis Group in the Department of Informatics at the University of Bergen, Norway. Before that, I was a tenured assistant professor in the Computer Graphics and Visualization group at TU Delft (until 2026) and a tenure-track assistant professor in the Leiden Computational Biology Center (until 2020) and a Postdoctoral researcher in the Computer Graphics and Visualization group at TU Delft. I received my PhD from King Abdullah University of Science and Technology in 2013.
My research interests include Visualization and Visual Analytics, with a focus on high-dimensional data and bio-/medical applications. I lead the research and development of Cytosplore, an interactive visual analytics platform for cytometry data which is used by researchers world-wide. In 2019, I received the Dirk Bartz Prize for Visual Computing in Medicine for the Cytosplore project. In 2023, I was awarded with the inaugural VRVis Visual Computing Award for contributions to sustainable development with visual computing. Further, my research won best paper and honorable mention awards at IEEE VIS, VCBM and PacificVis. For more detail, please refer to my full cv.
If you are a master student looking for a possible theses topic, we have several open projects and possibilities within topics related to data visualization in general, visual analytics and biomedical visualization. The projects can range fundamental to very applied. If you are interested in a project in the direction of my research, please, contact me and we can discuss the possibilities.
Exploration and analysis of high-dimensional data are important tasks in many fields that produce large and complex data, like the financial sector, systems biology, or cultural heritage. Tailor-made visual analytics...
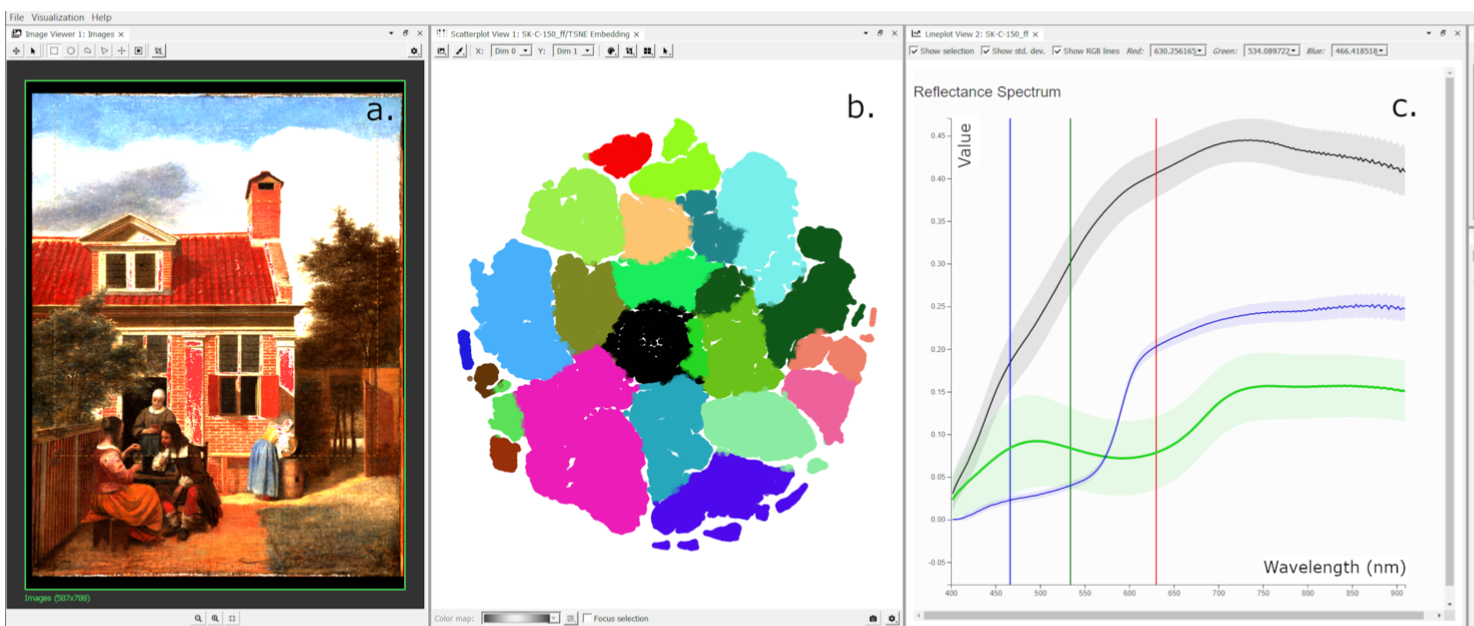
Reflectance Imaging Spectroscopy (RIS) is a hyperspectral imaging technique used for investigating the molecular composition of materials. It can help identify pigments used in a painting, which are relevant information...
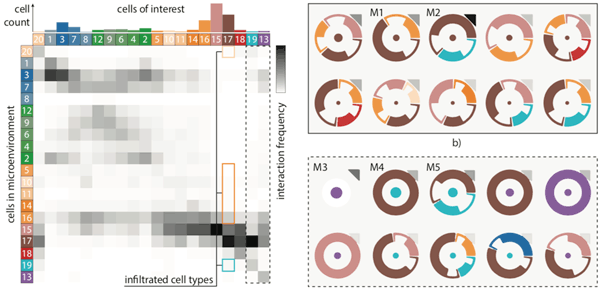
Tissue functionality is determined by the characteristics of tissue-resident cells and their interactions within their microenvironment. Imaging Mass Cytometry offers the opportunity to distinguish cell types with high precision and...
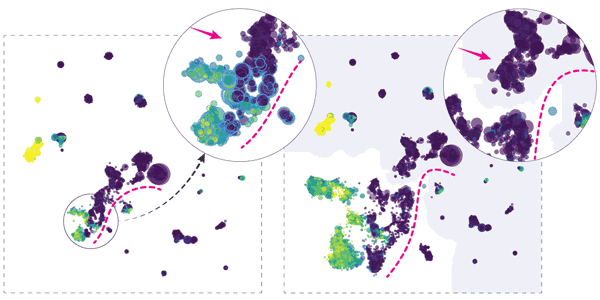
Hierarchical embeddings, such as HSNE, address critical visual and computational scalability issues of traditional techniques for dimensionality reduction. The improved scalability comes at the cost of the need for increased...

The primary motor cortex (M1) is essential for voluntary fine-motor control and is functionally conserved across mammals1. Here, using high-throughput transcriptomic and epigenomic profiling of more than 450,000 single nuclei...
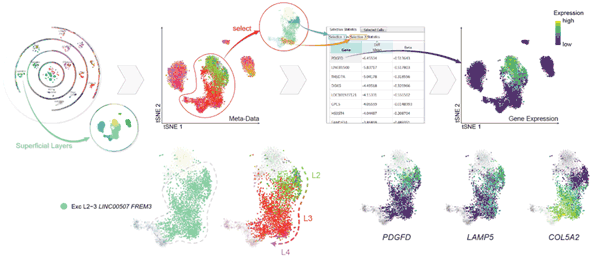
Elucidating the cellular architecture of the human cerebral cortex is central to understanding our cognitive abilities and susceptibility to disease. Here we used single-nucleus RNA-sequencing analysis to perform a comprehensive...